TK2d is a progressive, genetic, and life‑threatening mitochondrial disease.1


It can develop at any point in life, but early onset is defined as TK2d symptom onset that occurs on or before the age of 12.1
TK2d is the result of nuclear DNA mutation in the TK2 gene.1,2
This genetic mutation causes defects in the mitochondrial matrix enzyme, TK2, which is responsible for phosphorylating deoxythymidine (dT) and deoxycytidine (dC).1‑5
These nucleosides are crucial in the production and maintenance of mitochondrial DNA (mtDNA), and therefore, energy metabolism.1‑5
Without these nucleosides muscle damage occurs.1‑4
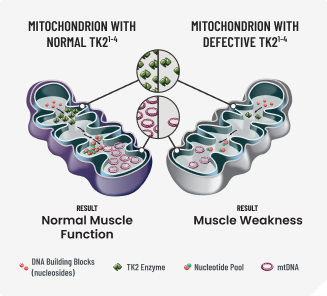
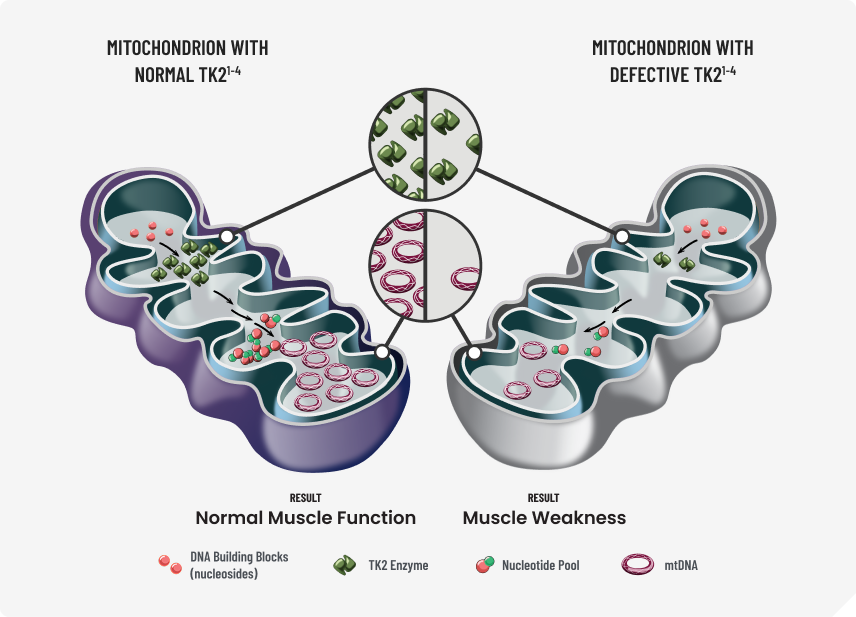
DNA=deoxyribonucleic acid; TK2d=thymidine kinase 2 deficiency.
Early-onset TK2d predominantly presents as muscle weakness.1
TK2d may impact the ability to walk, eat, and breathe independently. It is often fatal.1
Earlier onset typically results in more severe symptoms that progress faster and result in early death1,6
Once you identify red-flag symptoms, genetic testing is the fastest and only route to confirm a TK2d diagnosis.2,7
It is possible that a person first experiences symptoms as an infant or child but does not get diagnosed until after age 12 due to diagnostic delays. Because of this, it's possible that even those diagnosed as adults may have early-onset TK2d if their symptoms began on or before 12 years old.3,8
Conduct genetic testing early to help your patients seek the care they need.3,9
The TK2 gene is NOT included in all genetic tests. When you suspect TK2d, order testing inclusive of TK2 to definitively diagnose a patient.6,9
3 COMMON GENETIC TESTS TO DIAGNOSE TK2d:6
Whole-exome sequencing/whole-genome sequencing
PREFERRED METHOD
Multi-gene panels that include TK2
Single-gene testing for TK2
A definitive diagnosis is the first step toward treatment for early-onset TK2d.10
Some patients may need additional genetic testing.6,9
PATIENTS WHO HAVE:2,6,10,11
- Undergone genetic tests that did not include TK2
- Received negative or inconclusive results on neuromuscular panels or other genetic tests
- Received unconfirmed diagnoses of diseases that mimic TK2d (eg, neuromuscular disorders, Pompe disease, muscular dystrophy)
- Not yet received a diagnosis
Order genetic testing inclusive of the TK2 gene.
References
- Garone C, Taylor RW, Nascimento A, et al. Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet. 2018;55(8):515-521. doi:10.1136/jmedgenet-2017-105012
- Thymidine kinase 2 deficiency. National Organization for Rare Disorders. Updated March 1, 2026. Accessed March 1, 2026. https://rarediseases.org/rare-diseases/thymidine-kinase-2-deficiency/
- Berardo A, Domínguez-González C, Engelstad K, Hirano M. Advances in thymidine kinase 2 deficiency: clinical aspects, translational progress, and emerging therapies. J Neuromuscul Dis. 2022;9(2):225-235. doi:10.3233/JND-210786
- Lopez-Gomez C, Levy RJ, Sanchez-Quintero MJ, et al. Deoxycytidine and deoxythymidine treatment for thymidine kinase 2 deficiency. Ann Neurol. 2017;81(5):641-652. doi:10.1002/ana.24922
- Lopez-Gomez C, Hewan H, Sierra C, et al. Bioavailability and cytosolic kinases modulate response to deoxynucleoside therapy in TK2 deficiency. EBioMedicine. 2019;46:356-367. doi:10.1016/j.ebiom.2019.07.037
- Wang J, El-Hattab AW, Wong LJC. TK2-related mitochondrial DNA maintenance defect, myopathic form. In: Adam MP, Feldman J, Mirzaa GM, et al, eds. GeneReviews®. [Internet]. Seattle (WA): University of Washington, Seattle; December 6, 2012.
- Ng KWP, Chin HL, Chin AXY, Goh DLM. Using gene panels in the diagnosis of neuromuscular disorders: a mini-review. Front Neurol. 2022;13:997551. doi:10.3389/fneur.2022.997551
- Manini A, Meneri M, Rodolico C, et al. Case report: thymidine kinase 2 (TK2) deficiency: a novel mutation associated with childhood-onset mitochondrial myopathy and atypical progression. Front Neurol. 2022;13:857279. doi:10.3389/fneur.2022.857279
- Domínguez-González C, Madruga-Garrido M, Hirano M, et al. Collaborative model for diagnosis and treatment of very rare diseases: experience in Spain with thymidine kinase 2 deficiency. Orphanet J Rare Dis. 2021;16(1):407. doi:10.1186/s13023-021-02030-w
- Nicolau S, Milone M, Liewluck T. Guidelines for genetic testing of muscle and neuromuscular junction disorders. Muscle Nerve. 2021;64(3):255-269. doi:10.1002/mus.27337
- Grier J, Hirano M, Karaa A, Shepard E, Thompson JLP. Diagnostic odyssey of patients with mitochondrial disease: results of a survey. Neurol Genet. 2018;4(2):e230. doi:10.1212/NXG.0000000000000230